







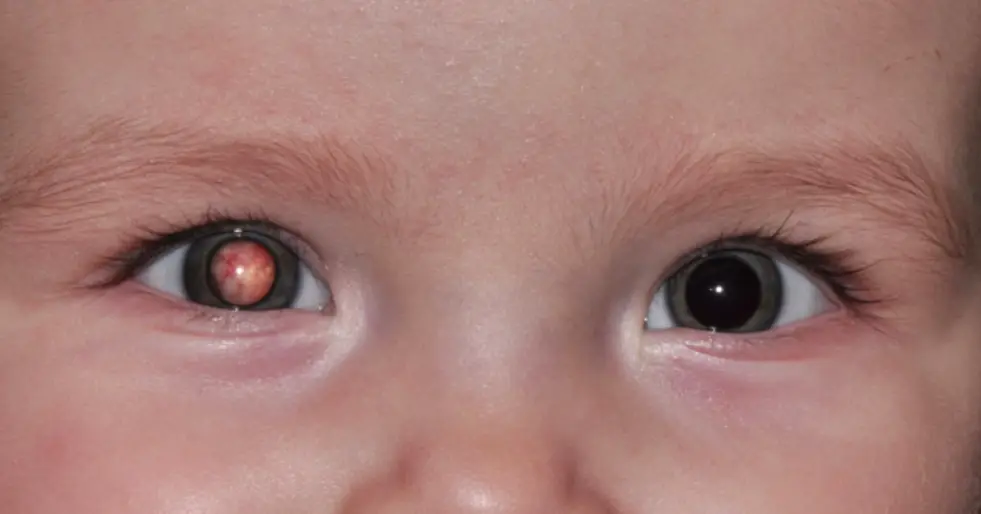
O retinoblastoma é o tumor intraocular maligno mais comum da infância, responsável por cerca de 3% das neoplasias pediátricas. Apesar de raro, seu impacto é profundo: ameaça a vida, a visão e traz sérias implicações psicossociais. O diagnóstico precoce é crucial para garantir altas taxas de cura (>90%) e maior chance de preservar o olho e a visão.
Nos últimos anos, avanços em diagnóstico molecular e terapêuticas conservadoras revolucionaram o manejo. Este artigo revisa, de forma objetiva, o que todo estudante e médico precisa saber sobre retinoblastoma.
Definição
O retinoblastoma é um tumor maligno que se origina das células precursoras da retina em desenvolvimento. Estas células, que normalmente se diferenciariam em fotorreceptores e outras células retinianas, sofrem transformação maligna devido a alterações genéticas específicas.
Este tumor representa uma emergência oftalmológica e oncológica, pois, se não tratado, pode crescer e invadir estruturas adjacentes, inclusive o nervo óptico, levando à cegueira, disseminação para o sistema nervoso central e metástases sistêmicas.
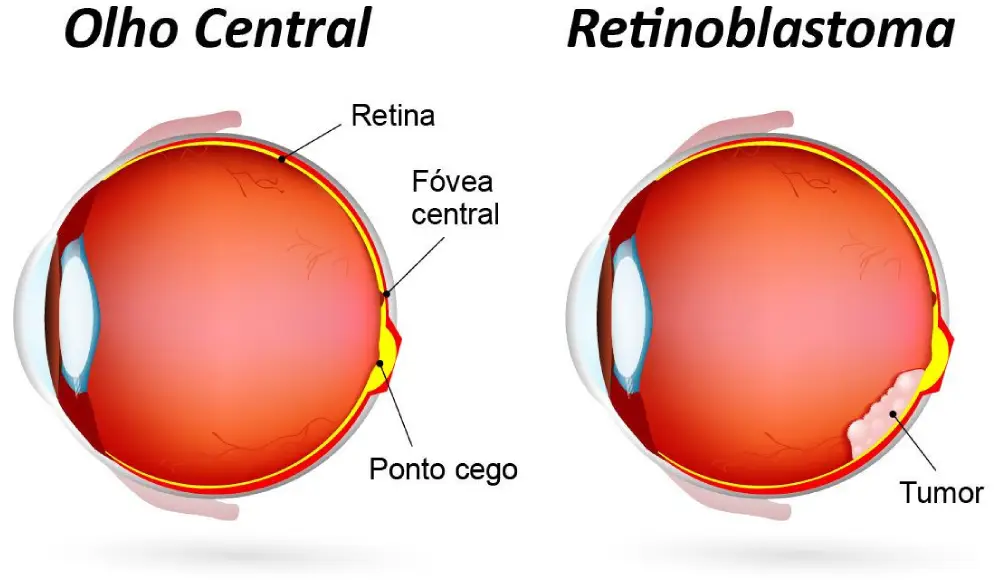
Etiologia
A causa fundamental do retinoblastoma reside na inativação bialélica (perda de função de ambas as cópias) do gene supressor de tumor RB1. A proteína codificada por este gene, conhecida como proteína do retinoblastoma (pRB), desempenha papel crucial como reguladora negativa do ciclo celular.
Quando ambos os alelos do gene RB1 são inativados, a proteína pRB funcional não é produzida ou é disfuncional, impulsionando a célula a proliferar de maneira descontrolada, culminando na formação do tumor.
O retinoblastoma classifica-se em duas formas principais com base na origem da mutação no gene RB1:
- Retinoblastoma Hereditário (Germinativo): Corresponde a 40-45% dos casos. A primeira mutação no RB1 está presente em todas as células do corpo (germinativa), podendo ser herdada ou surgir de novo. Caracteriza-se por:
- Doença frequentemente bilateral e/ou multifocal.
- Diagnóstico mais precoce (mediana de 12 meses).
- Risco aumentado de tumores secundários (osteossarcomas, pinealoblastoma – configurando a síndrome do retinoblastoma trilateral).
- Transmissão autossômica dominante com alta penetrância (cerca de 90%).
- Retinoblastoma Não Hereditário (Esporádico Somático): Corresponde a 55-60% dos casos. Ambas as mutações no RB1 ocorrem somaticamente em uma única célula da retina. Caracteriza-se por:
- Doença geralmente unilateral e unifocal.
- Diagnóstico um pouco mais tardio (mediana de 24 meses).
- Sem risco aumentado de tumores secundários.
- Não é transmitido hereditariamente.
Em raros casos (cerca de 2%), o retinoblastoma pode se desenvolver por mecanismos independentes do RB1, como a amplificação do oncogene MYCN.
Epidemiologia
Incidência, Prevalência, Distribuição Geográfica
A incidência global do retinoblastoma é estimada em aproximadamente 1 caso a cada 15.000-20.000 nascidos vivos, resultando em cerca de 8.000 novos casos diagnosticados anualmente em todo o mundo. No Brasil, um estudo populacional abrangente publicado em 2022 encontrou uma incidência de 2,23 casos por milhão para a faixa etária de 0-14 anos. Especificamente para a faixa etária de maior risco, de 0 a 4 anos, a incidência no Brasil foi de 7,02 casos por milhão.
O retinoblastoma ocorre em crianças de todas as etnias e em todas as partes do mundo. No entanto, existe uma marcante variação regional no Brasil: a taxa de incidência para crianças de 0-4 anos foi de 11,26 por milhão na região Nordeste, contrastando com 5,25 por milhão na região Centro-Oeste.
Fatores de Risco
- Mutação germinativa no gene RB1: principal fator.
- História familiar positiva de retinoblastoma.
- Fatores socioeconômicos e geográficos: associados a diagnóstico tardio e pior prognóstico, não à mutação em si.
Fisiopatologia
A fisiopatologia do retinoblastoma inicia-se com a inativação bialélica do gene RB1 em uma célula precursora da retina, provavelmente um precursor de cone.
A proteína pRB funcional atua como um “freio” molecular, ligando-se a fatores de transcrição da família E2F e impedindo a progressão do ciclo celular da fase G1 para a S. Sem pRB funcional, os fatores E2F são liberados, promovendo a transcrição contínua de genes que impulsionam a proliferação celular descontrolada, culminando na formação do tumor.
Processos celulares e moleculares envolvidos incluem:
- Instabilidade Genômica: A perda da função da pRB compromete os mecanismos que mantêm a integridade do genoma, levando a um aumento da taxa de mutações e rearranjos cromossômicos.
- Alterações Genômicas Adicionais: Além da inativação do RB1, alterações frequentes incluem amplificação de oncogenes como MYCN, E2F3, DEK e MDM4, bem como alterações em genes supressores de tumor como CDH11 e p75NTR.
- Evasão da Apoptose: As células do retinoblastoma desenvolvem mecanismos para evitar a morte celular programada, como superexpressão de proteínas antiapoptóticas e ativação de vias de sinalização de sobrevivência.
- Angiogênese Tumoral: Para sustentar seu crescimento, as células tumorais secretam fatores pró-angiogênicos como o VEGF, estimulando a formação de novos vasos sanguíneos.
Progressão tumoral
Inicialmente pode haver retinoma (benigno), que evolui para malignidade com alterações adicionais e pode seguir:
- Crescimento Endofítico: O tumor cresce para dentro da cavidade vítrea, com possível semeadura vítrea.
- Crescimento Exofítico: O tumor cresce em direção ao espaço sub-retiniano, causando descolamento de retina.
- Crescimento Misto: Combinação de componentes endofíticos e exofíticos.
- Retinoblastoma Infiltrante Difuso: Forma menos comum onde o tumor cresce de maneira plana.
Quadro Clínico
Os dois sinais de apresentação mais frequentes do retinoblastoma são:
- Leucocoria (60-80% dos casos): Classicamente descrita como “reflexo do olho de gato” ou “pupila branca”. Ocorre devido à reflexão da luz diretamente da superfície esbranquiçada do tumor. Frequentemente notada pelos pais em fotografias tiradas com flash.
- Estrabismo (20-30% dos casos): Desalinhamento ocular que surge quando o tumor afeta a visão central ou causa descolamento de retina, levando à perda da capacidade de fixação do olho afetado.
Manifestações de alerta/gravidade (menos comuns e/ou mais tardias)
- Sinais inflamatórios: Olho vermelho, dor ocular e inflamação intraocular.
- Proptose: Abaulamento do globo ocular (sinal tardio que indica doença avançada).
- Buftalmia: Aumento do tamanho do globo ocular, geralmente devido a glaucoma secundário.
- Dor de cabeça, vômitos, irritabilidade: Possíveis sinais de hipertensão intracraniana por disseminação para o SNC.
Diagnóstico
Anamnese e Exame Clínico
Uma história clínica detalhada deve incluir:
- Início, duração e progressão dos sinais ou sintomas (leucocoria, estrabismo, olho vermelho).
- História familiar de retinoblastoma ou outros tipos de câncer relacionados.
- Revisão de fotografias anteriores da criança (podem revelar leucocoria precoce).
No exame físico, o teste do reflexo vermelho deve sempre ser realizado durante as visitas de puericultura, além daquele que é realizado na maternidade. Se for identificada alguma alteração no reflexo (reflexo esbranquiçado ou acinzentado pode ser sinal de um retinoblastoma) ou se houver alguma dificuldade de realizar o teste, a criança deverá ser encaminhada ao oftalmologista.
Exames de Imagem
- Ultrassonografia Ocular (Modos A e B): Frequentemente o primeiro realizado, detecta massa intraocular sólida e calcificações intralesionais, mede dimensões do tumor e avalia descolamento de retina associado.
- Ressonância Magnética (RM) Orbitária e Cerebral com Contraste: Método de escolhapara avaliação detalhada e estadiamento. Avalia invasão do nervo óptico, coroide, esclera e tecidos orbitários e detecta disseminação intracraniana.
- Tomografia Computadorizada (TC) Orbitária: Deve ser evitada, pois a radiação está associada à formação de outros tumores em pacientes com a forma hereditária. Considerada apenas quando RM não está disponível.
Exames Laboratoriais
- Testes Genéticos: Essenciais. Realizados no sangue periférico para identificar mutações germinativas no RB1 (confirma forma hereditária) e, se houver enucleação, no tecido tumoral.
- Biópsia Líquida do Humor Aquoso: Técnica emergente, analisa DNA tumoral livre no humor aquoso para detectar mutações (RB1, MYCN), monitorar resposta terapêutica e prognóstico.
- Biópsia Tumoral Direta: Geralmente contraindicada devido ao risco de disseminação.
- Análise do LCR e Biópsia de Medula Óssea: Indicados para estadiamento em suspeita de doença extraocular.
Confira mais resumos da Dra. Olyvia Spontan:
Resumo de Pneumonia: Epidemiologia, Fisiopatologia, Manifestações Clínicas, Diagnóstico e Mais!
Tratamento
Objetivo
O tratamento é multidisciplinar e individualizado, visando salvar a vida em primeiro lugar, preservar o globo ocular e a visão útil sempre que possível e minimizar complicações e efeitos tardios.
Tratamento Farmacológico
Quimioterapia Sistêmica (Quimiorredução): Reduz o volume tumoral para permitir tratamentos locais subsequentes.
Quimioterapia Locorregional:
- Quimioterapia Intra-arterial (QIA): Cateterização superseletiva da artéria oftálmica e infusão de agentes quimioterápicos. Tem altas taxas de salvamento ocular (>95% em casos selecionados).
- Quimioterapia Intravítrea (QIV): Injeção de quimioterápicos diretamente no vítreo. É direcionada especificamente para semeadura vítrea ativa ou recorrente e requer técnica meticulosa para evitar disseminação extraocular.
Terapias Focais (Consolidação)
Fotocoagulação a Laser/Termoterapia Transpupilar: Utiliza laser infravermelho para aquecer e destruir o tumor; ideal para tumores pequenos no polo posterior.
Crioterapia: Aplicação de frio extremo na superfície externa da esclera; adequada para tumores periféricos.
Braquiterapia: Colocação cirúrgica de placa radioativa (I-125, Ru-106) sobre a base do tumor; opção para tumores de tamanho médio ou recorrentes.
Tratamento Cirúrgico
Enucleação (Remoção do Globo Ocular): Indicada para tumores grandes, glaucoma neovascular, falha de tratamentos conservadores, ou suspeita de invasão extensa. Um implante orbitário é colocado para resultado cosmético.
Manejo de Suporte e Sintomático
Inclui manejo de efeitos colaterais da quimioterapia (mielossupressão com G-CSF, transfusões; ototoxicidade; nefrotoxicidade; neurotoxicidade), complicações da QIA/QIV e da radioterapia. Suporte psicossocial e reabilitação visual são cruciais.
Complicações e prognóstico
- Complicações: Perda visual, necessidade de prótese ocular, metástases, neoplasias secundárias (principalmente após radioterapia), desfiguração, dor ocular crônica.
- Prognóstico: Sobrevida >90% em centros especializados para doença intraocular. Pior em casos extraoculares ou com diagnóstico tardio.
Conclusão
O retinoblastoma é um paradigma na oncogenética e um desafio contínuo na oncologia pediátrica. A compreensão de sua base molecular no gene RB1 e a distinção entre formas hereditárias e não hereditárias são cruciais para o manejo clínico, que evoluiu para abordagens multimodais focadas na preservação da vida, do olho e da visão.
O diagnóstico precoce, impulsionado pelo reconhecimento de sinais como leucocoria e estrabismo e pela triagem com o “Teste do Olhinho”, é o fator mais crítico para um bom prognóstico.
Para médicos e futuros médicos, a vigilância, o conhecimento atualizado e uma abordagem multidisciplinar são essenciais para oferecer o melhor cuidado possível a crianças com retinoblastoma.
Sobre a autora

Olyvia Spontan é médica formada pela Universidade Tiradentes (UniT) em 2022, com formação sanduíche na UPAEP (Universidad Popular Autónoma del Estado de Puebla – México) – ano de 2016.
Atua como médica reguladora e tem experiência em regulação de urgências e regulação de leitos. Além disso, é entusiasta da tecnologia, aliada à IA para redação de textos médicos, dos quais hoje é revisora.






